Frequently Asked Questions (FAQ) / Help Page
Looking for a quick introduction to SimpleSynteny? See the flyer! (PDF 1.5MB)
More details are also available in the paper, available here, from Nucleic Acids Research (Open Access).- What are the browser requirements to run SimpleSynteny?
- What are the file format requirements to use SimpleSynteny?
- My genome is not annotated! How do I know which contigs to choose?
- Why am I limited to only 10 genomes and 60 genes per file?
- Why am I limited to only 10 contigs per genome?
- Why are file sizes limited to only 100MB?
- Do you have any demo files for me to practice with?
- Is there a difference if I use one gene target file vs. multiple?
- How can I see if flanking genes appear about my target(s) of interest?
- How is the 'Minimum Query Coverage Cutoff' calculated?
- What is 'Circular Genome Mode'?
- My gene has duplicate copies, how do I remove one?
- Why are my genes not mapping to my genome?
- Why do my genome/gene names appear like '???' with all question marks?
- How do I interpret my final figure?
- No lines are being drawn between my genomes!
- What is the difference between the shading styles?
- My gene names are covering each other!
- Why are my arrows showing up behind/below contigs?
- My arrows seem to be stacking on top of each other!
- There's too much or not enough space between my genome names and the first contig!
- I want to leave out gene names so I can insert them using another program!
- What is the 'Reorder Genomes' option doing?
- Can I customize the colors of my gene targets?
- Can I annotate things manually? I want to include additional knowledge (exon splicing, tandem repeats, etc.) from other sources.
- What is 'Advanced Mode'?
- What is a 'CMAP' file?
- Do you have any example CMAP files?
- How do I remove gene directions and just draw plain rectangles?
- Is code available to run SimpleSynteny locally?
- How do I cite SimpleSynteny if I use it in my work?
Basic Usage and File Requirements:
Step1 - Upload and Search Settings:
Step2 - Contig Editing:
Step3 - Figure Generation and Editing:
Other / Advanced Usage:
Basic Usage and File Requirements
What are the browser requirements to run SimpleSynteny?
SimpleSynteny requires JavaScript and a modern browser that supports HTML5 and AJAX to work.
What are the file format requirements to use SimpleSynteny?
Genome File Requirements:
- Files must be in standard FASTA format.
- File names should end with a '.fasta' or '.fa', extension.
- Genome files can only contain DNA sequences.
- There is currently a maximum limit of 10 genome files.
- Genome files are currently limited to having between 1 and 10 contig/supercontig sequences.
- ID lines should only contain your contig/supercontig names- we recommend only using letters, numbers, underscores, periods and dashes.
- ID lines may not end with a space character.
- By default, contigs are processed in order first-to-last and accordingly drawn from left-to-right.
- Individual files must not exceed 100MB in size.
Query (Gene/Protein) File Requirements:
- Files must be in standard FASTA format.
- File names should end with a '.fasta' or '.fa', extension.
- Query files may contain DNA or standard protein sequences.
- There is currently a maximum limit of 10 gene files.
- Each gene file is currently limited to a max of 60 gene sequences.
- Identical gene names appearing in the same file will have numbers appended (e.g. MyGene.1, MyGene.2, MyGene.3 etc.) and treated as separate genes.
- ID lines may not end with a space character.
- When using multiple gene files, gene names without identical spelling will be treated as separate genes (e.g. MyGene1 ≠ MyGene ≠ My-Gene).
- Individual files must not exceed 100MB in size.
My genome is not annotated! How do I know which contigs to choose?
We recommend trying out our Contig Finder tool! First upload your genome (must be DNA sequences in FASTA format), then paste your genes/proteins of interest into the text box (use FASTA format) and search for any contigs which may contain them. You can then select up to 10 contigs to export in a separate FASTA file which should work with the main SimpleSynteny program.
Why am I limited to only 10 genomes and 60 genes per file?
Our computational resources are currently limited. Hopefully, in the future we may be able to handle submissions with more genomes and/or genes. SimpleSynteny was designed for a more focused analysis, for example looking at a specific gene cluster/region, rather than whole-genome annotation. Before running SimpleSynteny you need to already have an idea of what genes and contigs (see 'how to choose contigs' below) you are focusing on.
Why am I limited to only 10 contigs per genome?
This is partly due to our limited computational resources, but also to keep images readable. A gene such as aldolase may appear many times in a genome and our testing shows that so many lines are drawn in such cases the image becomes difficult to interpret. Also, given the image dimension and DPI limits our server can currently handle, more than around 10 contigs can cause the image to scale so small the text is illegible.
Why are file sizes limited to only 100MB?
This is a hard limit set by our hosting service.
Do you have any demo files for me to practice with?
Yes! You can try out our Demo Mode page where we've already uploaded the files or you can download them here.
Step1 - Upload and Search Settings
Is there a difference if I use one gene target file vs. multiple?
Yes, the difference determines if SimpleSynteny is being used for discovery or visualization. The SimpleSynteny visualization pipeline does not explicitly score or evaluate syntenic relationships between targets. Accordingly, users should be aware of the difference between assigning a single gene target file to multiple genomes compared to using specific files for each genome.
If you assign one gene target file to multiple genomes, or to a genome from which the sequences do no originate- BLAST searches will be performning novel discovery. Such evaluations can be a fast and convenient first step for researchers to visually explore syntenic relationships but confirmation of results using additional tools is required. This is particularly true if you are comparing evolutionarily distant species!
If you assign a unique gene target file to each genome specifically- SimpleSynteny is working strictly as a visualization tool and assumes the orthologous relationship between targets has already been confirmed by you, the user.
How can I see if flanking genes appear about my target(s) of interest?
If you are interested in genes that flank a particular target of interest, you should include these sequences in the same gene target file. Names for sequences are assigned from the FASTA definition line so you may wish to use a particular naming convention to make these stand out.
How is the 'Minimum Query Coverage Cutoff' calculated?
Please note this has changed to ignore gaps within hits since the earlier beta version of the program!
Only query sequences with a percentage of basepairs/residues falling within significant BLAST hits at, or above, this value will appear in your results. Using the
full-length gene query sequence, we tile the hits or 'HSPs' (High-scoring Segment Pair) which map with the same strand/direction as the HSP with the highest bitscore. A simplified example is shown below:
Original Query Gene: 1 ACCACCTTGAACAATCC 17
Genome Contig Sequence: 1 AACACCTCTCTCTTAAACTTT 21
BLAST HIT 1:
Query 1 ACCACCT 7
| |||||
Sbjct 1 AACACCT 7
BLAST HIT 2:
Query 6 CTTGAACAAT 15
||| ||| |
Sbjct 12 CTTAAACTTT 21
Now we map the significant hits back to the original:
Original: ACCACCTTGAACAATCC
Hit1: A-CACCT
Hit2: CTT-AAC--T
Combined: ACCACCTTGAACAAT--
Coverage: 15/17 (88.24%)
Note how the gaps within BLAST hits are ignored when calculating the final coverage score. If the 'Minimum Query Coverage Cutoff' was set to 88% this gene would map, however, if it was set to 89% it would not. This feature is included to help avoid queries with only a small fragment mapping to a genome from cluttering up results. Setting the value to '1' will show any query with at least one significant hit in your results.
What is 'Circular Genome Mode'?
This mode can be useful when dealing with circular genomes from bacteria or mitochondria. When using this mode, Genome1 acts as a reference for the order that genes appear. All other genomes will then be rotated to maximize the number of genes which match this order. This mode requires that each genome has only one circular contig and the mode will be disabled if you upload a genome file with more than one sequence.
Step2 - Contig Editing
My gene has duplicate copies, how do I remove one?
In Step2, if your genes map, you should see an example figure followed by a list of your genes (grouped by contig) with yellow "REMOVE" buttons. Click the button by the gene you wish to remove to update your figure. Note, genes drawn left-to-right on the figure are listed in the same order top-to-bottom and bit scores generated by BLAST are provided to help in selecting which copy to remove.
Why are my genes not mapping to my genome?
If you are confident your genes are present in your genome, there may be low sequence homology shared between your query and contig sequences and you will need to adjust the 'BLAST Settings for Gene Matches' in Step1. You can either increase the BLAST 'E-value Threshold' (see here for an explanation on BLAST E-values) or try lowering the 'Minimum Sequence Identity Cutoff to Map Genes.' The latter may especially be need if using protein sequences for your gene queries since proteins with as low as ~30% sequence identity may still have conserved function.
Why do my genome/gene names appear like '???' with all question marks?
SimpleSynteny currently only supports ASCII-encoded characters. We hope to include support for languages with non-ASCII characters in the future.
Step3 - Figure Generation and Editing
How do I interpret my final figure?
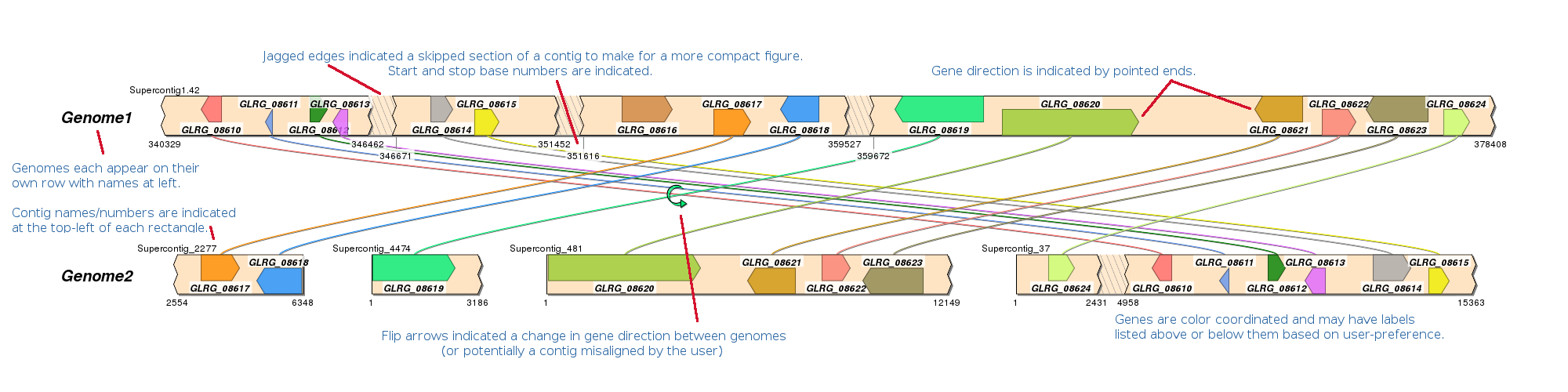
No lines are being drawn between my genomes!
Lines can only be drawn between genes with identical names. Make sure you mapped genes with the same name to each genome and we recommend using names with only letters, numbers and underscores.
What is the difference between the shading styles?
- The 'Project Full-Length Gene' option appears as follows: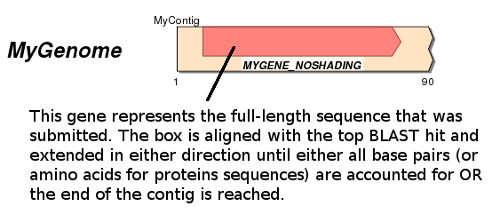
This mode does not detect introns and will assume they are not present!
- The 'Shade BLAST Region' style shows the projected full-length sequence as above, but darkens the region covering the highest scoring significant BLAST hit like in the figure below:
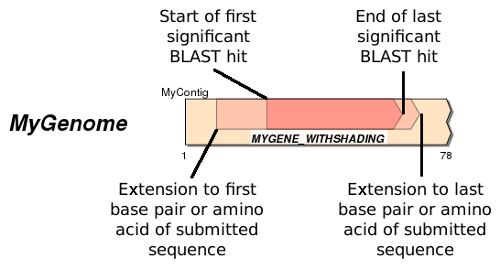
BLAST matches with 100% sequence identity will shade the entire box. Using genes from closely related taxa, this mode can be helpful to get a quick visual estimate of sequence identity. Currently, SimpleSynteny shades between multiple BLAST hits so intermediate gaps, indels, etc. will not be highlighted. Given enough interest this may be implemented in a future update.
- The 'Show BLAST Hits Only' (default) option will only show the region covering the highest scoring significant BLAST hit and will appear like the full-length gene version in the above figure (without shading). If you know there are introns present or if you feel the full-length gene projection is not accurately mapping your genes use this option.
My gene names are covering each other!
Genes are drawn from left-to-right on each contig so long gene labels may overlap. You can try reducing the font size by decreasing the "Gene Name Font Size" parameter or increasing the "Image Width" parameter under "User Settings" and redrawing your image. You may need to play with these numbers a few times until no more overlaps occur. Alternatively, you may want to edit your query genes file and use abbreviations for any long gene names. Because genes are often closely packed together on the same contig, we alternate gene labels to appear above and below their corresponding boxes to help fit more text in a small space.
Why are my arrows showing up behind/below contigs?
Try decreasing the "Arrow Spacing" value under "Genome Adjustments" so they pack closer together. You can also try increasing the "height" parameter under "User Settings" until all your arrows are visible.
My arrows seem to be stacking on top of each other!
Try increasing the "Arrow Spacing value under "Genome Adjustments" so they pack farther apart. Increasing the "height" parameter of the image may also help.
There's too much or not enough space between my genome names and the first contig!
Try changing the "Spacing After Genome Names" parameter under "User Settings" in Stage 3 until it looks correct. Increase the value to add space and decrease it to reduce it.
I want to leave out gene names so I can insert them using another program!
Try changing the "Remove Gene Names?" option to "Yes" under "Drawing Style" in Stage 3. Figures will then be drawn with empty white boxes so users can insert their own custom text/fonts with other software. Note, if you do not want the white boxes at all, also set the "Draw Box Around Gene Labels?" option to "No."
What is the 'Reorder Genomes' option doing?
If you have a many lines and arrows, sometimes figures can look quite busy. We offer two naive approaches to reorder your genomes (keeping the top genome in place) to try and produce a more readible figure. Note these can increase the amount of time it takes to generate your figure, particularly if you are comparing many genomes with many gene targets.
The 'Minimize Euclidean Line Distance' simply measures the total distance of all connections and trys to find the order with the shortest total distance. The 'Minimize Number of Flip Arrows' trys to find the order which generates the fewest total number of arrows.
Can I customize the colors of my gene targets?
Currently, the only way to customize the colors for gene targets is through the use of the local command line version of the program. We hope to add this functionality to the server in the future.
Other / Advanced Usage
Can I annotate things manually? I want to include additional knowledge (exon splicing, tandem repeats, etc.) from other sources.
Yes, you can manually insert new genes and adjust how boxes are shaded using 'Advanced Mode' (see next question) to include knowledge from outside programs. For example, if you have identified tandem repeats that BLAST may miss, you can insert these yourself.
What is 'Advanced Mode'?
Advanced Mode allows you to directly submit a CMAP file to fully customize your figure. You can either cut and paste lines from the CMAP files provided by regular SimpleSynteny output as a starting point, or write your own lines from scratch. This mode can also be used to manually insert introns/exons as separate gene entries.
What is a 'CMAP' file?
Please see the 'CMAP File Format' page for a detailed description.
Do you have any example CMAP files?
Yes, some CMAP files used to generate Fig. 2 from the paper are available here (.zip 13.8 KB). Included are CMAP files for 8 fungi along with protein sequences from a mating (MAT) gene cluster used to generate them. Most genomes are available online, please see the paper supplemental materials for details.
How do I remove gene directions and just draw plain rectangles?
You can remove directionality from a gene and display it as a plain rectangle by placing "==" (without quotes) on both sides of a gene name as in the following example:
Contig1 {1 [ 2193|3193 (1|1 == Ami1 == 2351|6351) 7685|8685 ] [ 12393|12393 (1|1 Cia30 ==> 756|756) 13148|13148 ] 14000}
In this case, the gene Ami1 would be drawn as a rectangle while Cia30 would be drawn with a right arrow like in this image:

Is code available to run SimpleSynteny locally?
SimpleSynteny scripts are available to run locally for larger jobs. Click here to download (v1.4.0 .ZIP 469kB).
How do I cite SimpleSynteny if I use it in my work?
Veltri, D., Malapi-Wight, M. and Crouch J.A. SimpleSynteny: a web-based tool for visualization of microsynteny across multiple species. Nucleic Acids Research 44(W1):W41-W45, 2016, doi:10.1093/nar/gkw330.
Still have a question you didn't find answered here?
Contact Dan Veltri at: dan.veltri@gmail.com with your questions. Please mention "SimpleSynteny" in the subject line.